Makoto Kurai MD (mkurai08@shinshu-u.ac.jp)1, 2, 3
Richard A. Robbins MD (rickrobbins@cox.net)1, 2, 5
Sekiya Koyama MD (syjskoyama@go.tvm.ne.jp) 4
Jun Amano MD, PhD (junamano@shinshu-u.ac.jp) 3
John M. Hayden PhD (John.Hayden2@va.gov)1
1Carl T. Hayden VA Medical Center, Phoenix, AZ, 85012, USA
2Arizona Respiratory Center, University of Arizona, Tucson, AZ, 85724, USA
3The Second Department of Surgery,
Shinshu University School of Medicine, Matsumoto 390-8621, Japan
4The Department of Pulmonary Internal Medicine,
National Hospital Organization Chushin Matsumoto Hospital, Matsumoto 390-0021, Japan
5Phoenix Pulmonary and Critical Care Research and Education Foundation, Gilbert, AZ 85295, USA
Abstract
Tiotropium bromide (Spiriva®) is used as a bronchodilator in chronic obstructive pulmonary disease (COPD). However, clinical evidence suggests that tiotropium bromide may improve COPD by mechanisms beyond bronchodilation. We hypothesized that tiotropium bromide may act as an anti-inflammatory agent by inhibiting monocyte chemotaxis, a process that plays an important role in the lung inflammation of COPD. To test this hypothesis monocytes were pretreated with tiotropium bromide prior to exposure to chemotactic agents and monocyte chemotactic activity (MCA) was evaluated with a blind chamber technique. Tiotropium bromide inhibited MCA in a dose- and time- dependent manner (respectively, p< 0.01) by directly acting on the monocyte. Acetylcholine (ACh) challenge increased MCA (p< 0.01), and tiotropium bromide effectively reduced (p< 0.01) the increase in MCA by ACh. The inhibition of MCA by tiotropium bromide was reversed by a muscarinic type 3 (M3)-muscarinic receptor antagonist (p< 0.01), and was not effected by an M2 receptor antagonist. Furthermore, a selective M3 receptor agonist, cevimeline, and Gq protein stimulator, Pasteurella multocida toxin, significantly increased MCA (P < 0.01), and tiotropium bromide pretreatment reduced (p< 0.01) the increase in MCA induced by these agents. These results suggest that tiotropium might regulate monocyte chemotaxis, in part, by interfering with M3-muscarinic receptor coupled Gq protein signal transduction. These results provide new insight that an anti-cholinergic therapeutic may provide anti-inflammatory action in the pulmonary system.
Introduction
Tiotropium bromide is a novel long-acting, inhaled, anticholinergic agent that is used as a treatment for chronic obstructive pulmonary disease (COPD). It has reported to have beneficial effects on the pulmonary function compared to other anticholinergic (short-acting) and beta-2 adrenergic agents. Although tiotropium predominantly functions as a bronchodilator, it reduces the development of acute exacerbations in COPD (1,2). These effects suggest that tiotropium might possess some function as an anti-inflammatory agent in addition to a bronchodilator (3-6). Tiotropium also has the potential to inhibit acetylcholine-induced proliferation of fibroblasts and myofibroblasts (7), further suggesting plausible beneficial influences on airway remodeling in COPD patients.
Acetylcholine (ACh) participates in the control of airway tone, which is an important factor contributing to the airway obstruction in the airway diseases (8). Anticholinergic agents effectively reverse the parasympathetic nerve stimulation and attenuate the smooth muscle contraction in the airway. This effect is especially important in regard to COPD because the parasympathetic nerve stimulation is augmented in the airway inflammation (9). Recent studies have demonstrated that ACh participates in the inflammatory processes through the release of chemotactic factors from alveolar macrophages and bronchial epithelial cells which subsequently promote inflammatory cell infiltration (10,11). Moreover, ACh treatment induces the proliferation of fibroblasts and myofibroblasts (7). Recently, it has also been reported that ACh synthesizing enzyme, choline acetyltransferase, is ubiquitously expressed in the airway cells (12), and that lung epithelial cells and pulmonary inflammatory cells can produce ACh and express functional muscarinic receptors (13). Thus, ACh may be involved in the airway inflammation, remodeling, and other pathophysiological phenomena acting in an autocrine and paracrine fashion (13,14).
In the present study, we evaluated tiotropium as an anti-inflammatory agent in monocyte chemotaxis. Moreover, we assessed key intracellular mechanisms that may regulate the inhibition of tiotropium-induced monocyte chemotaxis. We found that muscarinic receptor coupled G-protein signal transduction plays a role in the migration of monocyte. The results demonstrated that tiotropium inhibited the capability of monocytes to migrate to the chemotactic factor MCP-1, at least partly, by modulating muscarinic type 3 (M3) receptor coupled Gq protein signal transduction. These data suggest that tiotropium may play an anti-inflammatory role by inhibiting monocyte chemotaxis.
Materials and Methods
This study was conducted with the approval from the Research and Development and Institutional Review Board Committees of the Carl T. Hayden Veteran’s Affairs Medical Center, Phoenix, Arizona.
Purification of peripheral blood monocytes and the monocyte chemotaxis assay. Mononuclear cells for the chemotaxis assay were obtained from normal human volunteers by Ficoll-Hypaque density centrifugation to separate red blood cells and neutrophils from mononuclear cells (15). The enriched population of monocytes isolated by this method routinely display >98% viability as assessed by trypan blue exclusion (Sigma-Aldrich, St. Louis, MO). The cells were suspended in HBSS (Invitrogen, Carlsbad, CA) containing 2% bovine serum albumin (BSA, Sigma-Aldrich) at pH 7.5 to give a final concentration of 5 x 106 cells/ml. The suspension was then used for the monocyte chemotaxis assay.
The monocyte chemotaxis assay was performed by a 48-well microchemotaxis chamber (NeuroProbe, Cabin John, MD) as described previously (16). Briefly, 25 μL of a solution containing 100 ng/ml of recombinant human monocyte chemoattractant protein (MCP-1; Sigma-Aldrich) was placed into the lower wells, and a 10 μm thick polyvinylpyrrolidone-free polycarbonate filter (Nucleopore, Pleasanton, CA) with a pore size of 5 μm was placed over the bottom chamber. The concentration of MCP-1 used in this study was established previously (17). The silicon gasket and top pieces of the chamber were applied, and 50 μL of the cell suspension described above was placed into the top wells above the filter. The chambers were incubated in humidified air in 5% CO2 at 37° C for 90 min. After incubation, the chamber was disassembled, and non-migrated cells were wiped away from the filter. The filter was then immersed in methanol for 5 min, stained with Diff-Quik (American Scientific Product, McGraw Park, IL), and mounted on a glass slide. Cells that completely migrated through the filter were counted by using light microscopy (1000x) in at least 10 random high-power fields (HPF) per well.
Effects of tiotropium on MCP-1 - induced MCA. To evaluate the dose-dependent effects of tiotropium, monocytes were treated with 0, 0.01, 0.1, 1.0, 10, 100, 1000 nM for 30 min at 37º C in a humidified 5% CO2 atmosphere prior to MCA assay. After this interim, the cells were transferred to the chemotaxis chamber and exposed to the chemotactic agent for an additional 90 min in the environment described above.
To assess the time-dependent effects of tiotropium, monocytes were treated with 1000 nM tiotropium (maximal responsive dose) for varying times (0, 15, 30, 60, 90, 120 min) prior to MCA assay, and then they were transferred to the chemotaxis chamber and exposed to MCP-1 for additional 90 min.
The viability of monocytes after tiotropium exposure was evaluated by examining cell morphology and measuring lactate dehydrogenase (LDH) activity in the supernatant fluids. LDH activity was assessed by use of a commercially available kit (Tox-7; Sigma-Aldrich) according to the manufacture’s instructions. Dose- (up to 1000 nM) and time- (up to 3 hours) effects of tiotropium exposure on LDH activity in the supernatant fluids were not significant in these experiments (p > 0.3; data not shown). Based on observations of cellular morphology and LDH activity, no toxicity was observed with the concentration or time of exposure of tiotropium used in this study.
Effects of ACh on MCP-1 induced MCA and reversal by tiotropium. Since tiotropium inhibited MCP-1 induced MCA, we determined whether ACh directly interacted with the monocytes and increased their activity in vitro. Monocytes were treated with 100 μM ACh (Sigma-Aldrich) for 60 min at 37° C prior to transfer to the chemotaxis chamber and exposure to MCP-1 for an additional 90 min. The concentration of ACh used in this study was based on a dose that was established previously (8). Furthermore, to test the effects of tiotropium on MCA that was augmented by ACh, monocytes were treated with 1000 nM of tiotropium for 30 min at 37° C prior to ACh challenge. After this interim of exposure, the chemotaxis assays were performed as described above.
Effects of muscarinic receptor antagonists on MCP-1 induced MCA and abolishment of tiotropium effects. To determine which muscarinic receptor is involved with the regulation of MCA by tiotropium, gallamine (Sigma-Aldrich) an M2-receptor antagonist and 4-diphenylacetoxy-N-methylpiperidine methiodide (4-DAMP) (Sigma-Aldrich) an M3-receptor antagonist were used at the concentration of 300 µM, respectively, according to previous reports (10,11). Monocytes were exposed to these agents for 30 min before treatment with 1000 nM tiotropium for an additional 30 min. At this time the cells were transferred to the chemotaxis chamber as described above.
Effects of M3-receptor agonists on MCP-1 induced MCA. Because M3-receptor seemed to be involved with the ACh modulation, we evaluated whether tiotropium affected the reaction which was mediated through the M3-receptor agonist. A selective M3-receptor agonist, cevimeline HCl (EVOXAC®, Daiichi Sankyo, Inc., Parsippany, NJ), was used at the concentration of 300 μM. Monocytes were pretreated with 1000 nM tiotropium for 30 min at 37° C before the addition of cevimeline for an additional 30 min at 37° C prior to conducting the MCA assay.
Role of muscarinic receptor coupled Gq protein in MCA. Because M3 receptors are reported to couple with Gq proteins, we evaluated the effects of Gq protein stimulation on MCA. Pasteurella multocida toxin (PMT) was used as a Gq protein stimulator at the concentration of 100 ng/ml as previously reported (18,19). Monocytes were pretreated with 1000 nM tiotropium for 30 min before exposure to PMT for an addition 30 min at 37º C. The cells then were transferred to the microwell chambers and exposed to MCP-1 as described above.
Statistical Analyses. Unless stated otherwise all the results presented are expressed as means ± SEM from at least 3 individual experiments. Data were analyzed by one-way ANOVA, followed by selected post-hoc Neuman-Keuls multiple comparison tests. In selected experiments, Dunnett’s test was used to examine treatment effects compared to non-treated controls. In all cases, p < 0.05 was considered significant.
Results
Tiotropium inhibition of MCP-1 induced MCA. Tiotropium significantly inhibited MCP-1-induced MCA in a dose-dependent manner (Figure 1A, p< 0.005). The lowest dose to inhibit MCA was 10 nM (p< 0.05) and the greatest inhibition (~40%; p< 0.01) occurred with a dose of 1000 nM tiotropium. Tiotropium also significantly inhibited MCP-1-induced MCA in a time-dependent manner (Figure 1B, p< 0.001). MCA was inhibited significantly after 60 min (p< 0.01) and reached a plateau after 90 min of exposure to 1000 nM tiotropium.
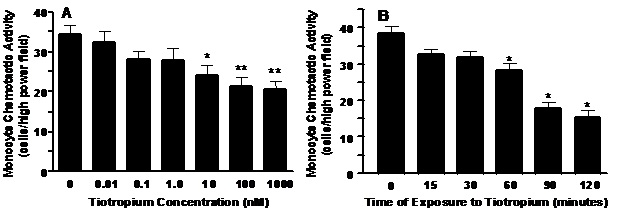
Figure 1. Panel A: A dose-dependent inhibition of monocyte chemoattractant protein (MCP-1) induced MCA by tiotropium. Monocytes were treated with tiotropium for 30 min before the MCA assay (n = 3). Chemotactic activity is on the ordinate and the concentration of tiotropium is on the abscissa. Values are expressed as means ± SEM. Dose effect by ANOVA (p < 0.005). *p< 0.05, **p< 0.01 means differ compared with non-tiotropium treated controls. Panel B: A time-dependent inhibition of MCP-1 induced MCA by tiotropium. Monocytes were treated with 1000 nM tiotropium under all conditions (n = 3). Chemotactic activity is on the ordinate and the duration of tiotropium exposure is on the abscissa. Values are expressed as means ± SEM. Time effect by ANOVA (p< 0.001). *p< 0.01 means differ compared to non-treated controls.
ACh augmentation of MCP-1 induced MCA. ACh challenge of the monocytes increased their MCA ~1.2-fold (Figure 2; p< 0.01). In addition, pretreatment of the monocytes with tiotropium inhibited (p< 0.01) MCA that was stimulated by ACh to a level that was lower than non-treated controls (Figure 2).
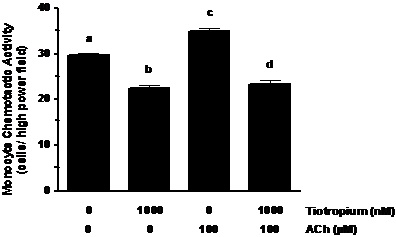
Figure 2. ACh augments MCA induced by MCP-1 exposure. Monocytes were treated with 1000 nM tiotropium for 30 min prior to exposure of 100 μM Ach (n = 3). Chemotactic activity is on the ordinate and the experimental groups are on the abscissa. Values are expressed as means ± SEM. a vs. b; a vs. c means differ (p< 0.01); c vs. d means differ (p < 0.001).
Muscaric receptor type 3 inhibitor reversed the effects of tiotropium. 4-DAMP pretreatment of the monocytes reversed the inhibitory effect of tiotropium on MCP-1 induced MCA (Figure 3). In contrast to 4-DAMP, the pretreatment of gallamine did not alter the reduction of MCA caused by anticholinergic treatment. As shown the figure 3, both gallamine and 4-DAMP that was administered individually did not affect the chemotaxis of monocytes when exposed to MCP-1.
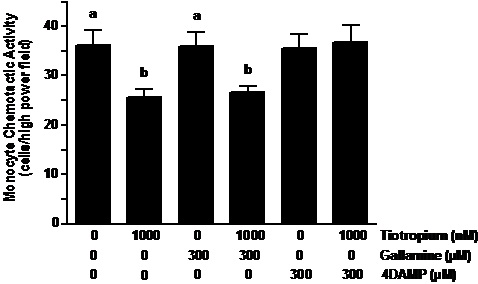
Figure 3. Muscarinic receptor type-3 inhibitor reversed the effects of tiotropium. Monocytes were pre-exposed to gallamine (M2 receptor inhibitor) and 4-diphenylacetoxy-N-methylpiperidine methiodide (4-DAMP; M3 receptor inhibitor) at the concentration of 300 µM for 30 min before treatment with 1000 nM tiotropium for an additional 30 min (n = 5). Chemotactic activity is on the ordinate and the experimental groups are on the abscissa. Values are expressed as means ± SEM. a vs. b means differ (p<0.01).
Tiotropium inhibition of a specific muscarinic type 3 receptor agonist induced MCA. Cevimeline (300 µM) treatment increased MCP-1 induced MCA as compared to nontreated controls (Figure 4, ~1.4-fold; p < 0.01). This effect was greater than that displayed by individual ACh treatment (Figure 2). In addition, tiotropium abolished the increase in MCA that was augmented by cevimeline treatment (Figure 4, p< 0.01).
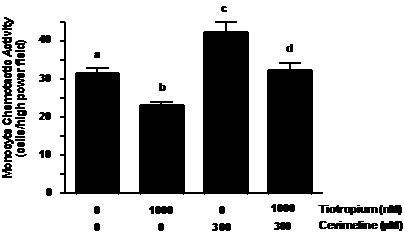
Figure 4. Tiotropium inhibited MCA induced by muscarinic type-3 receptor agonist treatment. Monocytes were treated with 1000 nM tiotropium for 30 minutes prior to exposure of 300 µM cevimeline for an additional 30 minutes (n = 4). Chemotactic activity is on the ordinate and the experimental groups are on the abscissa. Values are expressed as means ± SEM. a vs.b; a vs. c; c vs. d means differ (p< 0.01).
Tiotropium inhibition of Gq protein agonist induced MCA. MCA was significantly increased (1.3-fold; p< 0.01) in response to 100 ng/ml PMT as compared to non-treated controls (Figure 5). Similar to cevimeline, this effect was higher than that of ACh administered alone. In combination with PMT, tiotropium abolished the increase in MCA to a level that was similar to tiotropium treatment alone (Figure 5, p< 0.01).
Figure 5. Tiotropium inhibited MCA induced by Gq protein agonist treatment. Monocytes were treated with 1000 nM tiotropium for 30 min prior to exposure of 100 ng/ml Pasteurella multocida toxin (PMT) for an additional 30 min (n = 6). Chemotactic activity is on the ordinate and the both experimental groups are on the abscissa. Values are expressed as means ± SEM. a vs.b; a vs.c; c vs.d means differ (p< 0.001).
Discussion
In the present study, we demonstrated that tiotropium directly interacted with the monocyte and inhibited MCP-1 induced MCA. The exogenous addition of ACh increased MCP-1-induced MCA, and tiotropium reversed the increase in MCP-1 induced MCA by ACh. Interestingly, 4-DAMP, a M3-receptor antagonist, abolished the effect of tiotropium. Furthermore, cevimeline, a M3-receptor agonist, and PMT, a Gq protein stimulator, increased MCP-1 induced MCA, and tiotropium pretreatment reversed the increase in MCA by these agents. These data may suggest that tiotropium directly interacts with monocytes and inhibits their capability to migrate to chemotactic agents by interfering with M3 receptor coupled Gq protein signal transduction.
The initial concentrations used in this study demonstrated a reduction in MCA from tiotropium ranging from 10 through 1000 nM. The lower doses of tiotropium demonstrating a response in this study are within range to those affecting human lung fibroblast proliferation (7) and fibroblast differentiation (20). In order to elicit a robust effect on MCA, we opted to use the highest responsive dose (1000 nM) of tiotropium bromide throughout the study. At this level, tiotropium bromide was non-toxic and remained below the estimated maximum concentration of ~2 μM to be present at the lung epithelial lining fluid after inhalation of the drug (21).
Tiotropium has been shown to possess anti-inflammatory effects (1,2,9,13). Although the recruitment of peripheral blood monocytes to the lung is essential for innate lung immunity, it is also involved in the generation and propagation of an inflammatory response. The excessive migration of blood monocytes to the lung tissue can lead to increased number of alveolar macrophages (22,23), leading to the lung tissue injury via excessive elaboration of inflammatory cytokines, eicosanoids, proteolytic enzymes, and oxygen radicals (24-26). In this context, it might be important to suppress the excessive migration of monocytes to the site of inflammation during acute and chronic inflammation. In the present study, we demonstrated that tiotropium inhibited MCP-1 induced MCA. This result suggests that tiotropium may provide an anti-inflammatory action by inhibiting the monocytes capability to migrate to chemotactic agents that are produced at heightened levels under inflammatory conditions in the lung.
Recently, accumulating evidence demonstrates that acetylcholine and its synthesizing enzyme choline acetyltransferase (ChAT) are present not only in airway nerves, but also in various cells such as airway epithelial cells, endothelial cells, smooth muscle cells, lymphocytes, macrophages, mast cells, eosinophils and neutrophils (11). Furthermore, most inflammatory cells express functional muscarinic receptors (10,11,27,28). Muscarinic receptor agonists increase cytosolic Ca2+ and c-fos mRNA expression both in human T- and B- cell lines in an atropine-sensitive manner (29,30). These findings may suggest that ACh may regulate inflammatory processes in a paracrine and/or autocrine fashion(s) in inflammatory cells (29, 31-33). In the present study, ACh directly interacted with monocytes and stimulated their chemotactic activity. This observation is consistent with the above concept that ACh can regulate inflammatory processes. We demonstrated that tiotropium inhibited the increase in MCA which was induced by ACh. Moreover, tiotropium attenuated MCP-1 induced MCA in the absence of ACh. Several reports (12,29) suggest that ACh can be generated by ChAT that is localized in monocytes and regulate inflammatory processes in an autocrine and/or paracrine fashion(s). Based on these reports, we postulated that ACh can be generated within the monocyte in response to exterior stimuli and that tiotropium may modulate MCA that is augmented by intrinsic ACh.
Five different subtypes (M1-M5) of muscarinic receptor have been identified (34). Although muscarinic receptors are expressed in various inflammatory cells, the expression of each subtype seemed to be variable within each individual inflammatory cell. Although Fujii et al. (27) reported that all 5 classes of muscarinic receptors have been detected by RT-PCR in mononuclear cells, Bany et al. (35) reported that M2-M5 but not M1 mRNA were detected by RT-PCR. Furthermore, Costa et al. (36,37) and Hellstom-Lindahl et al. (38) reported that only M3-M5, but not other subtypes, were detected by RT-PCR. Therefore, the expression profile of muscarinic receptors seemed to be variable upon experimental conditions. Based on these reports, we investigated whether the effect of gallamine, a M2-receptor antagonist, and 4-DAMP, a M3-receptor antagonist, may alter the effect of tiotropium on MCA. We did not investigate other muscarinic antagonists because of the reports suggesting that the other muscarinic receptors were not strongly expressed in monocytes. Our results indicated that tiotropium may act by binding to M3 receptor. Profita et al. (39) also demonstrated that M3 receptors were observed in human blood monocytes by immunohistochemical detection. In addition, Sato et al. (11) suggested that ACh stimulated the bovine alveolar macrophages via M3 receptor to release factors promoting monocyte chemotactic activity. Furthermore, Fujii et al. (30) reported that the muscarinic receptor agonist, oxotremorine-M, increased c-fos mRNA expression in human T- and B- cell lines via the M3 muscarinic receptor because this effect could be blocked by 4-DAMP. Taken together, these reports suggest that ACh might be acting predominantly by M3 receptor regulation in mononuclear cells. Our results also demonstrate that 4-DAMP inhibited the effect of suppression of MCA by tiotropium. Therefore, it is feasible that ACh can be generated by monocytes themselves and act in an autocrine and paracrine manner via M3 muscarinic receptor interaction and that tiotropium may inhibit this reaction.
Recently, Han et al. (40) reported that a conformational change of the M3 receptors may occur in the immediate vicinity of the binding site of a G-protein coupled receptor (GPCR) activated by diffusible ligands such as the muscarinic agonist. Furthermore, several studies suggested that similar conformation changes occur in GPCR’s activated by diffusible ligands such as the beta 2-adrenergic receptor (41, 42). According to this context, the conformational change within the M3 receptor might occur by a muscarinic antagonist such as 4-DAMP in monocytes. It may be possible that a conformational change in the M3 receptor, as produced by 4-DAMP interaction, may alter the original binding site for tiotropium and thus reverse its affect on MCA.
Muscarinic receptors belong to the large family of G-protein coupled receptors (GPCR). The accumulating data has demonstrated that the “odd-numbered” muscarinic receptors (M1, M3, M5) couple preferentially to G-proteins of the Gq family, whereas the “even-numbered” receptors (M2, M4) prefer G-proteins belonging to the Gi/o family (10). Our result and several other reports demonstrate that the most important receptor on monocytes seem to be mediated via the M3 muscarinic receptor. Therefore, we examined the effect of a selective M3 muscarinic receptor agonist, cevimeline, and a selective Gq protein stimulator, PMT, on MCA. The results from these experiments demonstrated that both cevimeline and PMT were directly acting on monocytes and augmented MCP-1 induced MCA. In addition, tiotropium abolished the increase in MCA that were induced by these agents. These data may suggest that tiotropium may inhibit MCA via M3 muscarinic receptor and Gq protein signaling. Although it has been indicated that tiotropium may inhibit MCA via M3 receptor coupled Gq protein signaling, it remains unknown as to which intracellular signaling pathways beyond G-protein induction may be important in the regulation of MCA. The effects of tiotropium in the regulation of these intracellular mechanisms in monocytes remain as an important issue to be elucidated with future research.
In conclusion, tiotropium directly interacts with monocytes and inhibits their capability to migrate to chemotactic agents. The results in the present study provide new insight into mechanisms by which tiotropium may act as an anti-inflammatory agent in the pulmonary system. The reduction in monocyte migration may be one mechanism explaining the reduction in COPD exacerbations seen with tiotropium treatment in COPD.
Acknowledgements
Supported by a grant from Boehringer Ingelheim and the Phoenix Pulmonary and Critical Care Research and Education Foundation and the Department of Veterans Affairs. The contents do not represent the views of the Department of Veterans Affairs or the United States Government..
References
- Casaburi R, Mahler DA, Jones PW, Wanner A, San PG, ZuWallack RL, Menjoge SS, Serby CW, Witek T Jr. A long-term evaluation of once-daily inhaled tiotropium in chronic obstructive pulmonary disease. Eur Respir J 2002;19:217-224.
- Vincken W, van Noord JA, Greefhorst AP, Bantje TA, Kesten S, Korducki L, Cornelissen PJ. Improved health outcomes in patients with COPD during 1 yr's treatment with tiotropium. Eur Respir J 2002;19:209-216.
- Wollin L, Pieper MP. Tiotropium bromide exerts anti-inflammatory activity in a cigarette smoke mouse model of COPD. Pulm Pharmacol Ther 2010;23:345-54.
- Profita M, Bonanno A, Montalbano AM, Ferraro M, Siena L, Bruno A, Girbino S, Albano GD, Casarosa P, Pieper MP, Gjomarkaj M. Cigarette smoke extract activates human bronchial epithelial cells affecting non-neuronal cholinergic system signaling in vitro. Life Sci 2011;89:36-43.
- Profita M, Riccobono L, Montalbano AM, Bonanno A, Ferraro M, Albano GD, Gerbino S, Casarosa P, Pieper MP, Gjomarkaj M. In vitro anticholinergic drugs affect CD8+ peripheral blood T-cells apoptosis in COPD. Immunobiology 2012;217:345-53.
- Profita M, Bonanno A, Montalbano AM, Albano GD, Riccobono L, Siena L, Ferraro M, Casarosa P, Pieper MP, Gjomarkaj M. β(2) long-acting and anticholinergic drugs control TGF-β1-mediated neutrophilic inflammation in COPD. Biochim Biophys Acta 2012;1822:1079-89.
- Pieper MP, Chaudhary NI, Park JE. Acetylcholine-induced proliferation of fibroblasts and myofibroblasts in vitro is inhibited by tiotropium bromide. Life Sci 2007;80:2270-2273.
- Barnes NC, Evans J, Zakrzewski JT, Piper PJ, Costello JF. A critical evaluation of the clinical relevance of leukotrienes in bronchial asthma. Agents Actions 1989;Suppl 28:123-133.
- Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to emphysema. N Engl J Med 1984;311:421-425.
- Koyama S, Sato E, Nomura H, Kubo K, Nagai S, Izumi T. Acetylcholine and substance P stimulate bronchial epithelial cells to release eosinophil chemotactic activity. J Appl Physiol 1998;84:1528-1534.
- Sato E, Koyama S, Okubo Y, Kubo K, Sekiguchi M. Acetylcholine stimulates alveolar macrophages to release inflammatory cell chemotactic activity. Am J Physiol 1998;274:L970-979.
- Wessler IK, Kirkpatrick CJ. The non-neuronal cholinergic system: an emerging drug target in the airways. Pulm Pharmacol Ther 2001;14:423-434.
- Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respir Res 2006;7:73.
- Racke K, Juergens UR, Matthiesen S. Control by cholinergic mechanisms. Eur J Pharmacol 2006;533:57-68.
- Boyum A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of nonnuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest Suppl 1968;97:77-89.
- Harvath L, Falk W, Leonard EJ. Rapid quantitation of neutrophil chemotaxis: use of a polyvinylpyrrolidone-free polycarbonate membrane in a multiwell assembly. J Immunol Methods 1980;37:39-45.
- Sato E, Simpson KL, Grisham MB, Koyama S, Robbins RA. Effects of reactive oxygen and nitrogen metabolites on MCP-1-induced monocyte chemotactic activity in vitro. Am J Physiol 1999;277:L543-549.
- Bagley KC, Abdelwahab SF, Tuskan RG, Lewis GK. Pasteurella multocida toxin activates human monocyte-derived and murine bone marrow-derived dendritic cells in vitro but suppresses antibody production in vivo. Infect Immun 2005;73:413-421.
- Essler M, Hermann K, Amano M, Kaibuchi K, Heesemann J, Weber PC, Aepfelbacher M. Pasteurella multocida toxin increases endothelial permeability via Rho kinase and myosin light chain phosphatase. J Immunol 1998;161:5640-5646.
- Disse B, Speck GA, Rominger KL, Witek TJ Jr, Hammer R. Tiotropium (Spiriva): mechanistical considerations and clinical profile in obstructive lung disease. Life Sci 1999;64:457-464.
- Matthiesen S, Bahulayan A, Kempkens S, Haag S, Fuhrmann M, Stichnote C, Juergens UR, Racke K. Muscarinic receptors mediate stimulation of human lung fibroblast proliferation. Am J Respir Cell Mol Biol 2006;35:621-627.
- Adamson IY, Bowden DH. Role of monocytes and interstitial cells in the generation of alveolar macrophages II. Kinetic studies after carbon loading. Lab Invest 1980;42:518-524.
- Blusse van Oud Alblas A, van der Linden-Schrever B, Van Furth R. Origin and kinetics of pulmonary macrophages during an inflammatory reaction induced by intra-alveolar administration of aerosolized heat-killed BCG. Am Rev Respir Dis 1983;128:276-281.
- Fels AO, Cohn ZA. The alveolar macrophage. J Appl Physiol 1986;60:353-369.
- Nathan CF. Secretory products of macrophages. J Clin Invest 1987;79:319-326.
- Sibille Y, Reynolds HY. Macrophages and polymorphonuclear neutrophils in lung defense and injury. Am Rev Respir Dis 1990;141:471-501.
- Fujii T, Kawashima K. An independent non-neuronal cholinergic system in lymphocytes. Jpn J Pharmacol 2001;85:11-15.
- Strom TB, Deisseroth A, Morganroth J, Carpenter CB, Merrill JP. Alteration of the cytotoxic action of sensitized lymphocytes by cholinergic agents and activators of adenylate cyclase. Proc Natl Acad Sci USA 1972;69:2995-2999.
- Fujii T, Kawashima K. Ca2+ oscillation and c-fos gene expression induced via muscarinic acetylcholine receptor in human T- and B-cell lines. Naunyn Schmiedebergs Arch Pharmacol 2000;362:14-21.
- Fujii T, Kawashima K. Calcium signaling and c-Fos gene expression via M3 muscarinic acetylcholine receptors in human T- and B-cells. Jpn J Pharmacol 2000;84:124-132.
- Kawashima K, Fujii T. Extraneuronal cholinergic system in lymphocytes. Pharmacol Ther 2000;86:29-48.
- Kirkpatrick CJ, Bittinger F, Nozadze K, Wessler I. Expression and function of the non-neuronal cholinergic system in endothelial cells. Life Sci 2003;72:2111-2116.
- Wessler I, Kilbinger H, Bittinger F, Unger R, Kirkpatrick CJ. The non-neuronal cholinergic system in humans: expression, function and pathophysiology. Life Sci 2003;72:2055-2061.
- Caulfield MP Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev 1998;50:279-290.
- Bany U, Ryzewski J, Maslinski W. Relative amounts of mRNA encoding four subtypes of muscarinic receptors (m2-m5) in human peripheral blood mononuclear cells. J Neuroimmunol 1999;97:191-195.
- Costa P, Auger CB, Traver DJ, Costa LG. Identification of m3, m4 and m5 subtypes of muscarinic receptor mRNA in human blood mononuclear cells. J Neuroimmunol 1995;60:45-51.
- Costa P, Traver DJ, Auger CB, Costa LG. Expression of cholinergic muscarinic receptor subtypes mRNA in rat blood mononuclear cells. Immunopharmacology 1994;28:113-123.
- Hellstrom-Lindahl E, Nordberg A. Muscarinic receptor subtypes in subpopulations of human blood mononuclear cells as analyzed by RT-PCR technique. J Neuroimmunol 1996;68:139-144.
- Profita M, Giorgi RD, Sala A, Bonanno A, Riccobono L, Mirabella F, Gjomarkaj M, Bonsignore G, Bousquet J,Vignola AM. Muscarinic receptors, leukotriene B4 production and neutrophilic inflammation in COPD patients. Allergy 2005;60:1361-1369.
- Han SJ, Hamdan FF, Kim SK, Jacobson KA, Bloodworth LM, Li B, Wess J. Identification of an agonist-induced conformational change occurring adjacent to the ligand-binding pocket of the M(3) muscarinic acetylcholine receptor. J Biol Chem 2005;280:34849-34858.
- Ghanouni P, Steenhuis JJ, Farrens DL, Kobilka BK. Agonist-induced conformational changes in the G-protein-coupling domain of the beta 2 adrenergic receptor. Proc Natl Acad Sci U S A 2001;98:5997-6002.
- Jensen AD, Guarnieri F, Rasmussen SG, Asmar F, Ballesteros JA, Gether U. Agonist-induced conformational changes at the cytoplasmic side of transmembrane segment 6 in the beta 2 adrenergic receptor mapped by site-selective fluorescent labeling. J Biol Chem 2001;276:9279-9290.
Conflict of Interest Statement: M.K., S.R., R.A.R., S.K., and J.A.H. do not have any financial relationship with a commercial entity that has an interest in the subject of this manuscript. J.A.H. received a research grant ($49,900) from Boehringer-Ingelheim (2006-7) to conduct this study.
Reference as: Kurai M, Robbins RA, Koyama S, Amano J, Hayden JM. Tiotropium bromide inhibits human monocyte chemotaxis. Southwest J Pulm Crit Care 2012;5:86-99. (Click here for a PDF version of the manuscript).