32 Year Old Man with “Community-Acquired” Pneumonia
Jill K. Gersh, M.D., MPH1, Michelle K. Haas MD2,3,4
1Department of Medicine, University of Colorado Denver, Aurora, CO; 2Denver Health Medical Center, Denver, CO; 3Denver Metro Tuberculosis Clinic, Denver, CO; 4Division of Infectious Diseases, Department of Medicine, University of Colorado Denver, Aurora, CO
Corresponding author: Jill Gersh, M.D., MPH Phone: 303-602-5052 Fax: 303-602-5055. Email: JILL.GERSH@UCDENVER.EDU
All authors declare they have no conflicts of interest to disclose.
Abstract
Background: Community-acquired pneumonia is a common reason for hospital admission; however underlying pathogens vary depending on host immunity and circulating pathogens in the community.
Case Summary: A 32 year old man from Malawi presented with community-acquired pneumonia. After failing outpatient management, he was admitted and found to have underlying HIV disease. His diagnostic work up was initially inconclusive for M. tuberculosis (TB) and thus his diagnostic evaluation and treatment focused on other etiologies. He was ultimately diagnosed with TB after an invasive procedure and had a rapid clinical response after initiating TB treatment.
Conclusion: Both failure to recognize that TB can present with a syndrome similar to bacterial pneumonia and over-reliance on diagnostic testing delayed the diagnosis of TB. Delays in diagnosis contributed to substantial morbidity and risked nosocomial transmission. Despite declining incidence in the US, providers should remain cognizant of diagnostic limitations for TB disease and have a low threshold for empiric treatment.
Introduction
Community-acquired pneumonia (CAP) is a common reason for presentation to care. The epidemiology of CAP can vary depending on the patient’s community of origin and underlying co-morbidities (1). We present a case of a 32 year old man who presented with CAP in whom his diagnosis was delayed due to failure to fully consider these factors.
Case
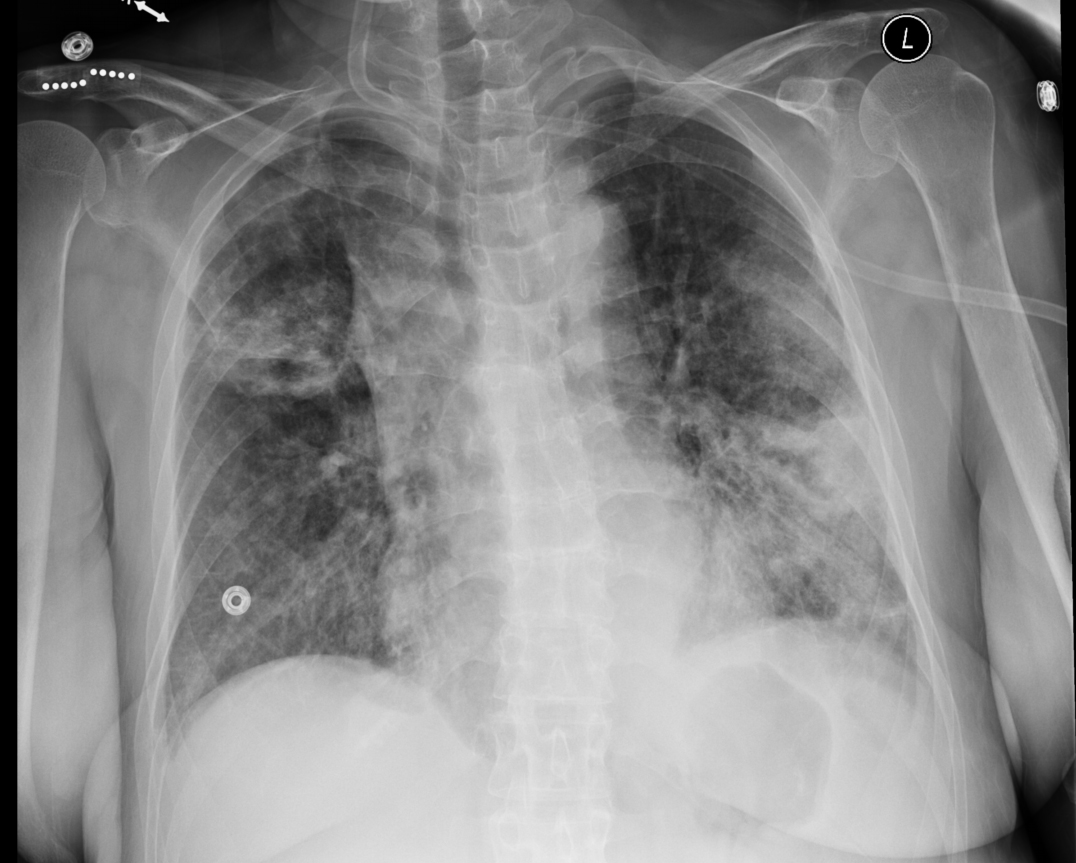
A 32 year old man from Malawi[1] presented to the emergency department (ED) with cough and dyspnea that failed to respond to a 5 day course of azithromycin. Chest radiography (CXR) was performed (Figure 1), demonstrating right middle lobe consolidation with ipsilateral hilar lymphadenopathy (LAD).
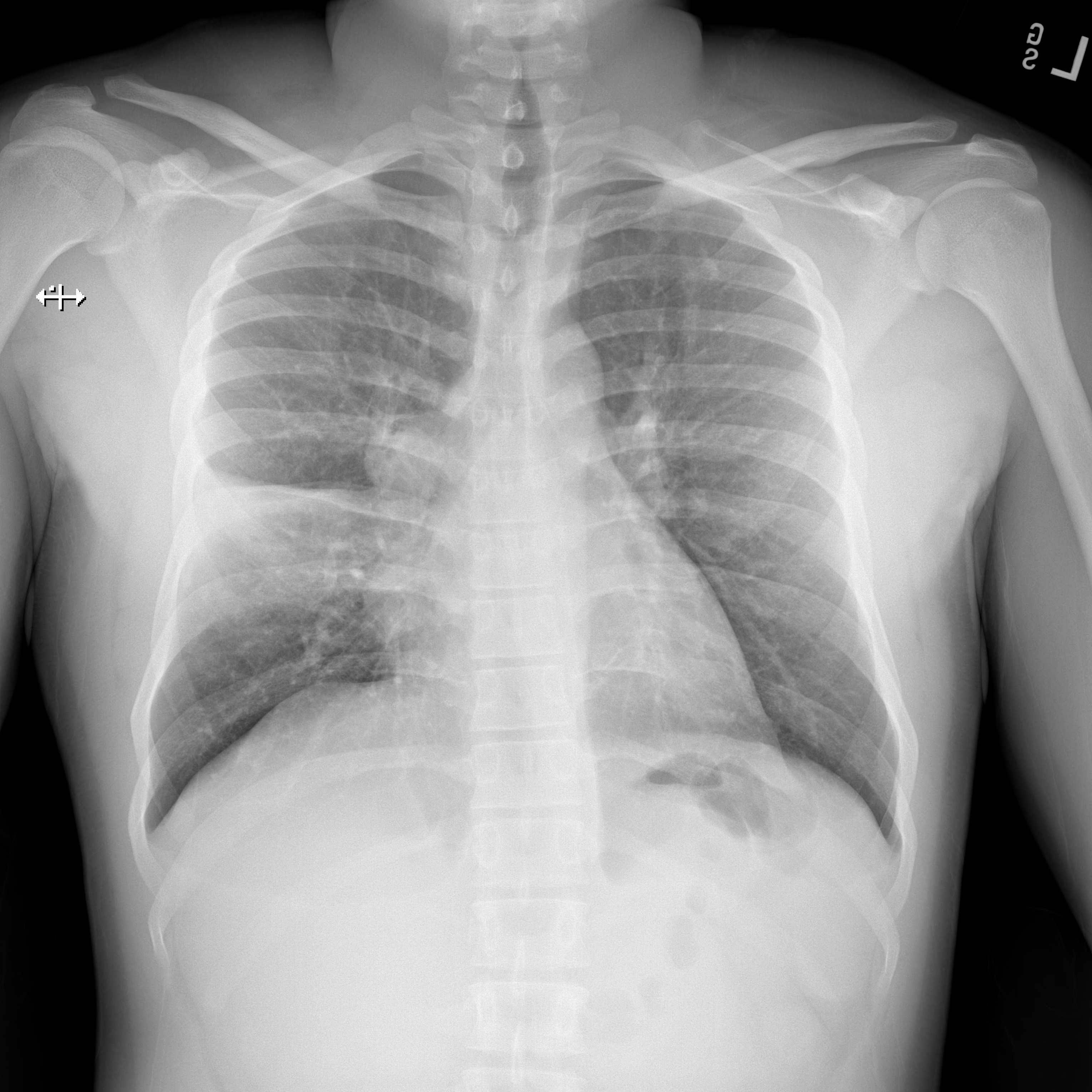
Figure 1. PA view of the chest demonstrating right middle lobe consolidation and ipsilateral hilar lymphadenopathy at the time of his first ED presentation and approximately 10 days into his illness.
He was diagnosed with CAP and discharged with a 7 day course of amoxicillin-clavulanate. His symptoms progressed with fevers, and weight loss. He presented for the second time to the ED and repeat CXR showed worsening right-sided hilar LAD and right middle lobe consolidation (Figure 2).
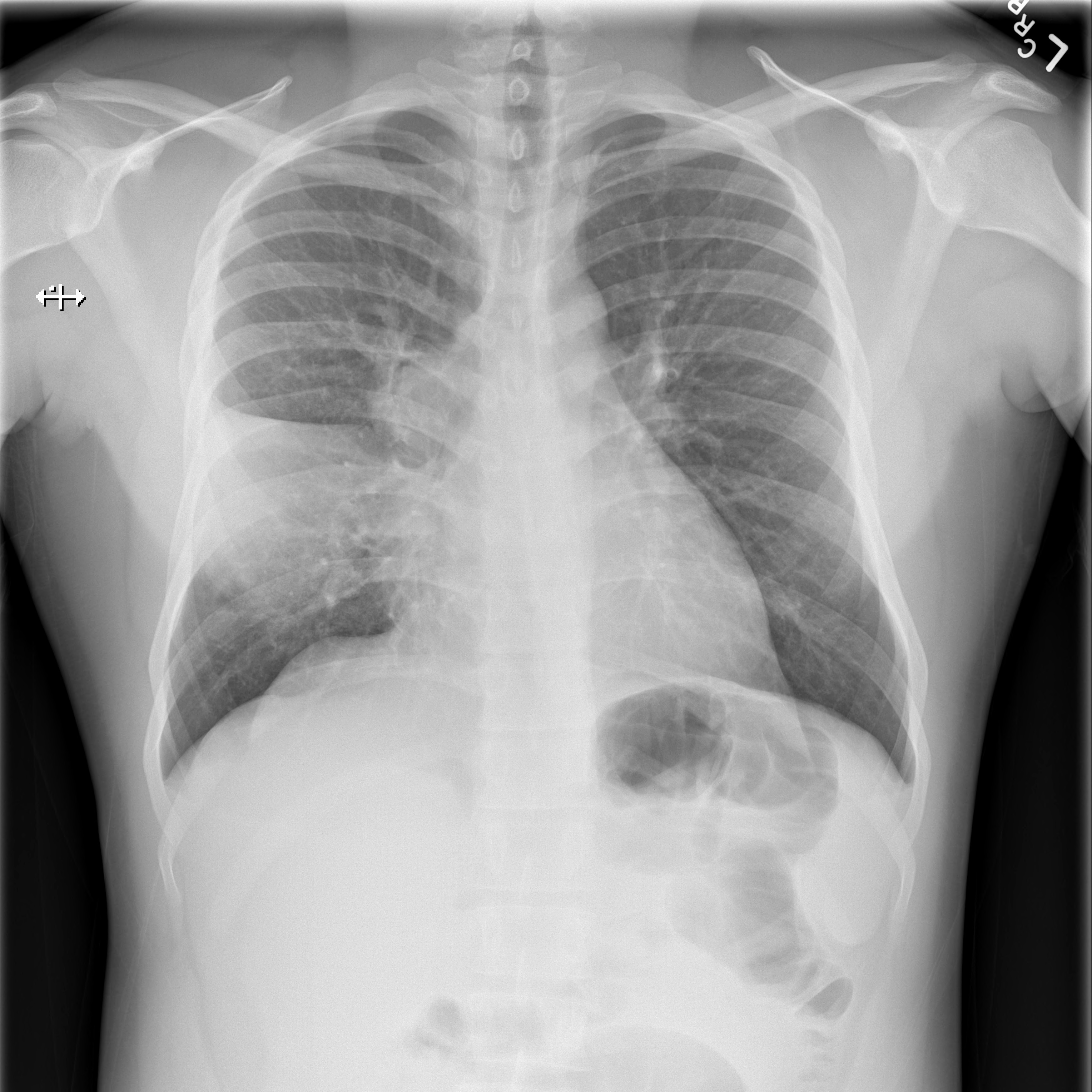
Figure 2. PA view of the chest demonstrating worsening of right middle lobe consolidation and right sided hilar lymphadenopathy at the time of admission to the hospital and approximately 17 days into his illness.
A rapid HIV test was positive and his CD4 count was 60 cells/µL. He was admitted and started on ceftriaxone, azithromycin and trimethoprim-sulfamethoxazole daily. He was placed on respiratory isolation and three sputum samples for acid-fast bacilli (AFB) smear and culture were collected, all of which were AFB smear negative. He then underwent bronchoscopy and his bronchoalveolar lavage smear was negative for AFB. His tuberculin skin test (TST) was negative as was an interferon gamma release assay (IGRA). He was then removed from respiratory isolation.
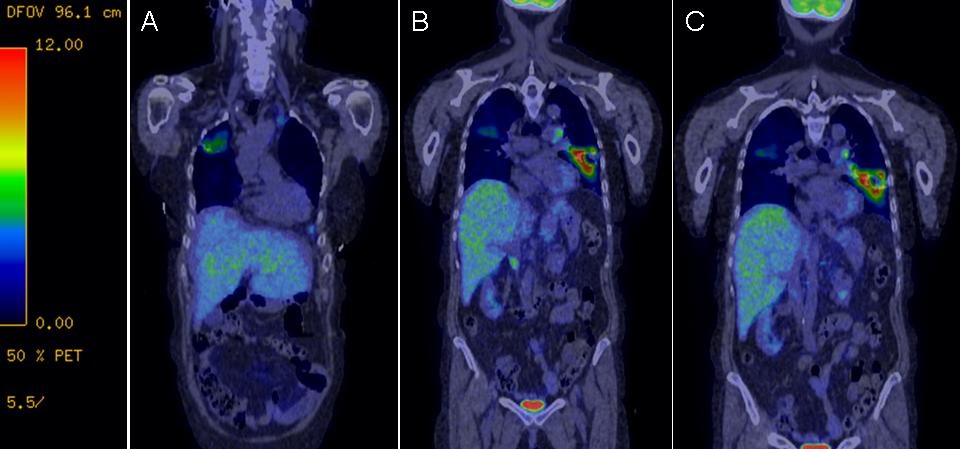
He continued to worsen with daily fevers as high as 43ºC while antimicrobial coverage was broadened to vancomycin and cefepime. He eventually underwent mediastinoscopy and lymph node (LN) biopsy. The following day LN tissue was positive for AFB and probe identified Mycobacterium tuberculosis (TB). Twenty-nine days after his initial presentation and 15 days into his hospitalization he was started on anti-tuberculosis therapy with isoniazid, rifampin, pyrazinamide and ethambutol. His cough improved within 2 days, his fevers were gone by day 4 and he was discharged. All sputum cultures grew TB. Antiretroviral therapy was initiated five weeks into his TB treatment. He had an excellent clinical and radiographic response (Figure 3) and completed 9 months of TB treatment.
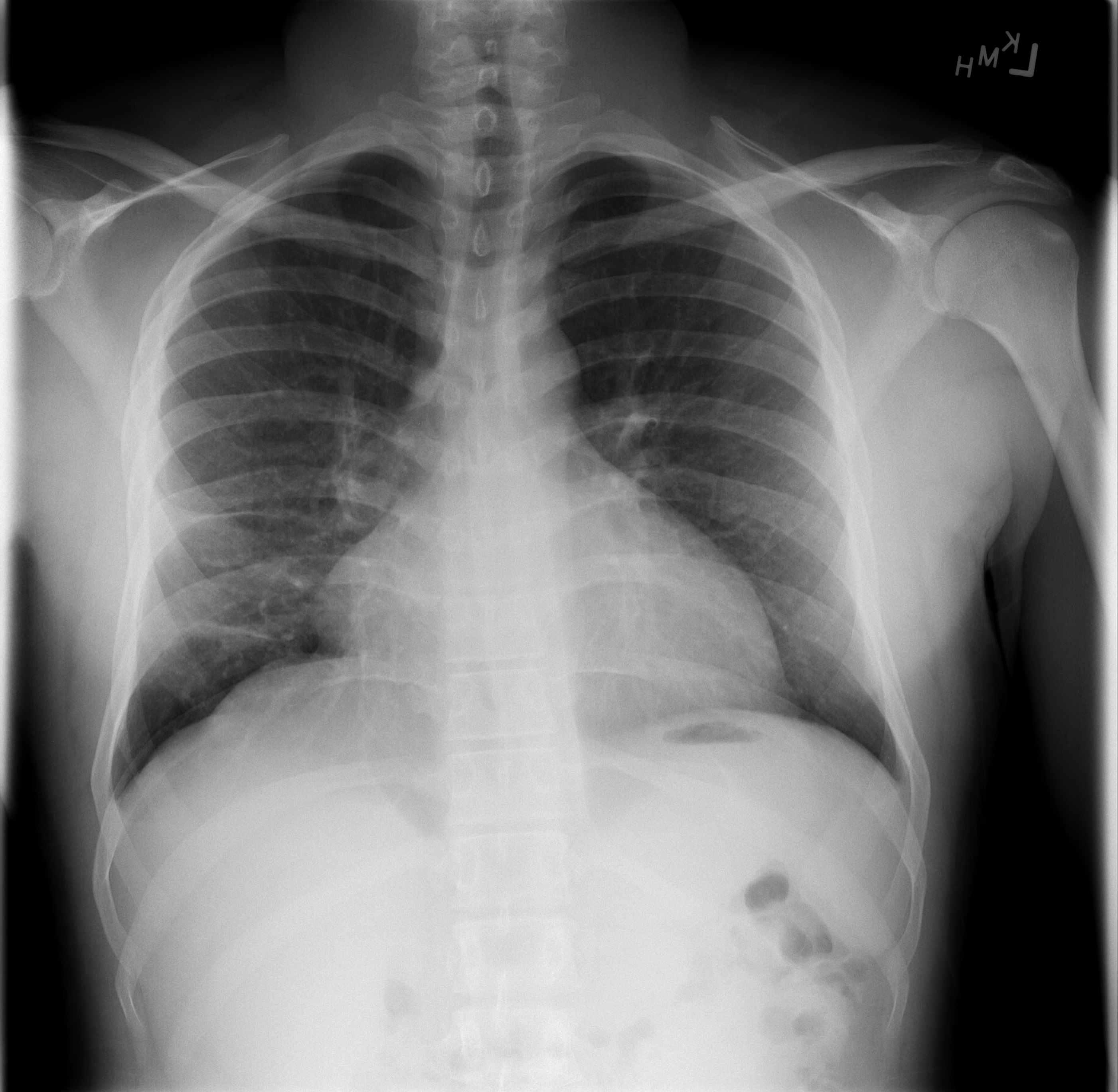
Figure 3. PA view of the chest after 9 months of treatment for M. tuberculosis. Noted here is resolution of right sided hilar lymphadenopathy and resolution of his right middle lobe consolidation with some residual scarring noted. Sputum culture converted at 2 months.
Diagnosis: Pulmonary tuberculosis.
Discussion
TB is the leading cause of death among HIV-infected individuals globally and the leading cause of morbidity in HIV-infected individuals (2). TB can present as an acute pneumonia with rapid progression of disease including sepsis and respiratory failure. Cough may not be a prominent feature and may be of less than two weeks duration. Additional signs and symptoms include fevers, night sweats, weight loss, hepatosplenomegaly, and lymphadenopathy. Individuals with CD4 counts < 100 cells/µL are more likely to present with disseminated disease and less likely to have cavitary disease. HIV-infected patients are also more likely to present with AFB smear negative disease even when severely ill (3). Chest radiograph findings vary from normal appearing films to hilar lymphadenopathy, diffuse infiltrates, and lobar consolidation.
TST and IGRAs are often negative and serve as poor screening tools for active disease. Up to 25% of individuals may have a negative TST or IGRA while having active disease, particularly HIV-infected individuals with advanced immunodeficiency (4). A negative result should never lower the clinical suspicion for active TB.
Delays in TB treatment are a major contributor to excess mortality in HIV-infected patients (2). The importance of early empiric treatment in HIV-infected individuals cannot be overstated. The World Health Organization (WHO) published guidelines in 2007 for the management of HIV-infected individuals suspected of having TB (5). While WHO guidelines are developed for low resource settings, these guidelines have relevance in the U.S. when managing patients with HIV who have lived or traveled to areas with a high burden of TB.
The failure to recognize that his clinical syndrome of CAP included TB as the underlying pathogen led to delayed treatment, prolonged hospitalization and risked nosocomial transmission. One unintended consequence of the success of our TB control programs may be the growing lack of clinical experience with TB among our providers. More broadly, how much of what we do as U.S. healthcare providers is because we can, and instead of what we should? Imagine if he couldn't get a mediastinoscopy and biopsy. Is it possible that his treatment course would have been improved by a lack of these resources? We would do well to learn from our colleagues practicing in resource limited settings where prescribing empiric TB treatment and assessing for a clinical response is standard of care. In this patient’s case, less really would have been more.
References
- Nyamande K, Lalloo UG, John M. TB presenting as community-acquired pneumonia in a setting of high TB incidence and high HIV prevalence. Int J Tuberc Lung Dis. 2007;11(12):1308-13. [PubMed]
- Wong EB, Omar T, Setlhako GJ, et al. Causes of death on antiretroviral therapy: a post-mortem study from South Africa. PloS one. 2012;7(10):e47542. [CrossRef] [PubMed]
- Elliott AM, Halwiindi B, Hayes RJ, Luo N, Tembo G, Machiels L, Bem C, Steenbergen G, Pobee JO, Nunn PP, et al. The impact of human immunodeficiency virus on presentation and diagnosis of tuberculosis in a cohort study in Zambia. J Trop Med Hyg. 1993;96(1):1-11. [PubMed]
- Cattamanchi A, Ssewenyana I, Davis JL, Huang L, Worodria W, den Boon S, Yoo S, Andama A, Hopewell PC, Cao H. Role of interferon-gamma release assays in the diagnosis of pulmonary tuberculosis in patients with advanced HIV infection. BMC Infect Dis. 2010;10:75. [CrossRef] [PubMed]
- Improving the diagnosis and treatment of smear-negative pulmonary and extrapulmonary tuberculosis among adults and adolescents: recommendations for HIV-prevelent and resource constrained settings. Geneva: World Health Organization;2007.
Acknowledgements
The authors wish to thank Carolyn Welch, MD for her thoughtful review of this case report.
Reference as: Gersh JK, Haas MK. 32 year old man with "community-acquired' pneumonia. Southwest J Pulm Crit Care. 2013;7(6):355-9. doi: http://dx.doi.org/10.13175/swjpcc173-13 PDF
 Post a Comment
Post a Comment