Makoto Kurai1,2,3
Richard A. Robbins1,2
Sekiya Koyama4
Jun Amano3
John M. Hayden1
1Carl T. Hayden Veterans Affairs Medical Center, Phoenix, Arizona 85012, 2Arizona Respiratory Center, University of Arizona, Tucson, Arizona 85724, 3Second Department of Surgery, Shinshu University School of Medicine, Matsumoto 390-8621, Japan, 4Department of Pulmonary Internal Medicine, National Hospital Organization Chushin Matsumoto Hospital, Matsumoto 399-0021, Japan
Abstract
Tiotropium, a long-acting anticholinergic, may improve chronic obstructive pulmonary disease (COPD) by mechanisms beyond bronchodilatation. We tested the hypothesis that tiotropium may act as an anti-inflammatory mediator by directly acting on and inhibiting human neutrophil chemotactic activity (NCA) that is promoted by acetylcholine (ACh) exposure. ACh treatment increased NCA in a dose dependent manner (p < 0.001) and tiotropium pretreatment reduced ACh stimulation (dose effect; 0 to 1000 nM; p < 0.001). Selective muscarinic receptor inhibitors demonstrated that subtype-3 (M3) receptor plays a role in NCA regulation. In addition, NCA that was stimulated by cevimeline (M3 agonist) and pasteurella multocida toxin (PMT, M3 coupled Gq agonist). However, the increased NCA to cevimeline and PMT was reduced by tiotropium pretreatment (p < 0.001). ACh treatment stimulated ERK-1/2 activation by promoting protein phosphorylation and tiotropium reduced this effect (p < 0.01). In addition, pretreatment of the cells with a specific MEK-1/2 kinase inhibitor reduced ACh stimulated NCA (p < 0.01). Together these results demonstrated that cholinergic stimulation of NCA is effectively inhibited by tiotropium and is governed by a mechanism involving M3 coupled Gq signaling and downstream ERK signaling. This study further demonstrates that tiotropium may act as an anti-inflammatory agent in lung disease.
Abbreviation List
- Ach – acetylcholine
- ANOVA – analysis of variance
- AS - complement activated serum
- BCA - bicinchoninic acid
- ChAT - choline acetyltranferase
- COPD – chronic obstructive pulmonary disease
- ERK - extracellular-signal-regulated kinases
- GAPDH - glyceraldehyde-3-phosphate dehydrogenase
- LPS – lipopolysaccharide
- M3 – muscarinic subtype 3 receptor
- MEK - mitogen-activated protein/extracellular signal-regulated kinase
- NCA – neutrophil chemotactic activity
- PMT - pasteurella multocida toxin
- rhIL-8 - recombinant human interleukin-8
- RIPA - radioimmunoprecipitaion assay
- SEM – standard error of mean
- TBST - tris-buffered saline and tween 20
Introduction
Anticholinergic therapy has been regarded as a first choice bronchodilator in the management of stable chronic obstructive pulmonary disease (COPD) (1). The agents included within this class of therapeutics effectively reverse the stimulation of parasympathetic produced acetylcholine (ACh) on smooth muscle airway contraction. Parasympathetic activity is increased with airway inflammation, and in regards to COPD, is an important mechanism because vagal tone appears to be one of the only reversible components of airflow restriction (2). Besides bronchoconstriction, ACh may also be involved in airway remodeling and other pathophysiogical mechanisms that are important in the propagation of lung disease (1,3-8). Recently it has been suggested that ACh may be expressed in the lung independent of a parasympathetic mechanism. In support of this notion, ACh synthesizing enzyme (choline acetyl transferase) has been found to be ubiquitously expressed in the airways, pulmonary epithelial cells, and immune cells such as neutrophils and monocytes (9-12). In addition, these cells also appear to express functional muscarinic receptors (9-11). Interestingly, the expression and function of certain muscarinic receptors in neutrophils may be increased in COPD (13), thus suggesting increase bioactivity associated with enhanced lung inflammation. We and others have previously demonstrated that ACh may also stimulate resident lung cells to release chemotactic factors and subsequently these factors can induce pro-inflammatory chemotaxis indirectly in vitro (3,4,8,9).
It has been recently reported that outcomes of COPD are improved by inhalation of cholinergic inhibitors, and tiotropium (tiotropium bromide, Spiriva®; Boehringer Ingelheim, Ingelheim, Germany) demonstrates the greatest improvements in COPD because of its long-acting, once daily administered, anticholinergic capability (1). Although tiotropium predominantly functions as a bronchodilator, it has also been shown to inhibit ACh-induced proliferation of fibroblasts and myofibroblasts (16), and inhibit the release of chemotactic factors from cultured lung epithelial cells, fibroblasts and alveolar macrophages in vitro (3,4). Taken together these results suggest a plausible beneficial role of tiotropium on airway remodeling and action as an anti-inflammatory agent in chronic airway disease.
We have previously reported that supernatants from macrophages that were treated with tiotropium prior to a challenge with lipopolysaccharide (LPS) greatly reduced the subsequent stimulation of NCA and this result did not occur by inhibited release of chemotactic factors (17). Based on these results, we postulated that tiotropium from the test media may actually passively diffuse through the pores of the filter that separates the chambers of the microchemotaxis unit and possibly interact directly with the neutrophils. In this study, we tested the hypothesis that tiotropium may act as an anti-inflammatory agent by directly interacting on neutrophils and inhibiting their chemotactic capability.
It has been well established that infiltration of neutrophils and the modulation of their activity play an important role in propagating and governing inflammation in a variety of lung diseases such as COPD (18). In addition, muscarinic receptor G-protein coupled signal transduction (19) and downstream ERK-1/2 activity (20-22) may also play an important regulatory role in controlling the migration of neutrophils. In this study, we further demonstrate that tiotropium may inhibit NCA, in part, through the regulation of muscarinic receptor coupled Gq-protein and ERK-1/2 mediated signal transduction (Figure 1).

Figure 1. Putative mechanism of tiotropium effect on neutrophil chemotaxis. Acetylcholine (ACh) either exogenously released or acting in a paracrine fashion stimulates the muscarinic type 3 (M3) receptor. This subsequently activates Gq protein which activates extracellular-signal-regulated kinases (ERK) 1/2. ERK 1/2 translocates into the nucleus activating various transcription factors which result in cell migration. Tiotropium decreases chemotaxis by inhibiting the binding of ACh to the M3 receptor.
Methods
This study was conducted with the approval from the Research and Development and Institutional Review Board Committees of the Carl T. Hayden Veteran’s Affairs Medical Center, Phoenix, Arizona.
Purification of Human Blood Neutrophils and Experimental Models
Human primary neutrophils were isolated and purified from heparinized plasma obtained from normal healthy individuals by the method of Böyum (23). The purified neutrophils were exposed to ACh (sodium acetylcholine, Sigma-Aldrich) up to 60 min prior to chemotaxis. For most experiments, cells were also pretreated with the various factors listed below for 30 min prior before selected agonist treatment. Inclusive of these agents are tiotropium bromide (Boehringer-Ingelheim); muscarinic (M) receptor antagonists including pirenzepine dihydrochloride (M1; Sigma-Aldrich); gallamine trithiodide M2 (M2; Sigma-Aldrich); 4-diphenylacetoxy-N-methylpiperidine methiodide (4-DAMP; M3; Sigma-Aldrich); M3 receptor agonist cevimeline HCl (EVOXAC®, Daiichi Sankyo, Inc., Parsippany, NJ); selective G-protein agonists(Gq, Pasteurella multocida toxin [PMT], EMD Biosciences Inc., San Diego, CA, Go, mastoparan, Biomol International, Plymouth Meeting, PA) and a specific mitogen-activated protein/extracellular signal-regulated kinase (MEK)-1/2 inhibitor (U0126; Sigma-Aldrich).
Neutrophil Chemotaxis Analysis
The chemotaxis assay was performed in a 48-well microchemotaxis chamber (NeuroProbe Inc., Cabin John, MD) using previously described methods (14). Either recombinant human interleukin-8 (rhIL-8, Sigma-Aldrich) or complement activated serum (AS) were used as the chemoattractants. Neutrophil viability was assessed and not altered by tiotropium.
Western Blot Analysis
The examination of corresponding regulation of extracellular signal-regulated kinase (ERK)-1/2 proteins by ACh and tiotropium in neutrophils was performed by Western Blot analyses. Both phosphorylated (p) and total (t) ERK-1/2 proteins were examined. Rabbit monoclonal antibodies directed against human pERK-1/2, tERK-1/2, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) proteins were purchased from Cell Signaling Technology (Beverly, MA).
For the ACh time-course experiment, neutrophils (1 x 107) were treated with 100 μM ACh for period ranging from 0 to 60 min of exposure. After establishing the maximal time effect (~15-20 min), subsequent experiments were conducted examining the effect of a 30 min tiotropium (100 nM) pretreatment on ACh challenged ERK-1/2 protein expression.
Neutrophils were lysed with ice-cold radioimmunoprecipitaion assay (RIPA) buffer including a proteolytic inhibitor cocktail (Santa Cruz Biotechnology, Santa Cruz, CA) as per the manufacturer’s instructions. Total protein concentration of the lysates was determined by the bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, Rockford IL). Protein concentrations were then adjusted to 40 µg in a standardized volume before addition of 2x sample buffer (Invitrogen, Grand Island, NY) and heating for 5 min at 85˚C. Cell proteins were then separated by electrophoresis on 4-20% tris-glycine acrylamide gels (Invitrogen, Grand Island, NY) and transferred to membranes (HCL-hybond, GE Healthcare, Piscataway, NJ) by electroblotting at 25 volts overnight at 4oC. The membranes were then pretreated with 1x tris-buffered saline and tween 20 (TBS-T) plus 5% non-fat dried milk for at least 2 hours at room temperature before exposure to the primary antibodies (1:2000) as per manufacturer’s suggestion overnight at 4oC. After subsequent washing with TBS-T a horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (1:2000) was added for at least 1 h at room temperature.
After multiple washings, the membranes were exposed to peroxidase substrate for enhanced chemiluminescence (Pierce ECL Western Blotting Substrate, Thermo Fisher Scientific, Rockford, IL) for 5 min. Membranes were wrapped and placed against autoradiograph film (Hyperfilm ECL; GE Healthcare, Piscataway, NJ) and developed (up to 30 min). The resulting protein bands were quantified by densitometry (Personal Densitometer SI, Image Quant ver. 5, Molecular Dynamics, GE Healthcare Biosciences Corp.).
Statistical Analyses
Unless stated otherwise data are means ± SEM resulting from at least 3 individual experiments. Data were analyzed by one-way ANOVA followed by selected post-hoc Neuman-Keuls tests. p < 0.05 was considered significant.
Results
Stimulation of neutrophil chemotactic activity by cholinergic challenge
Neutrophils were pretreated with varied concentrations of ACh ranging between 1-100 µM prior to exposure to two different chemotactic agents including rhIL-8 (500 ng/ml) and AS. As demonstrated in Figure 2, ACh treatment stimulated NCA in a dose dependent manner for both IL-8 and AS (p < 0.001). Similarly, at the 1 or 10 µM level ACh stimulated NCA when exposed to either IL-8 or AS, respectively. Moreover, the maximal level of stimulation by ACh was attained when the cells were treated with 100 µM ACh (Figure 2). As reported previously, this concentration of ACh provided maximal effects in other cell types (13,15). Beyond the dose effect studies, we also tested the effect of duration of ACh exposure (15 to 60 min) on NCA and found a significant stimulatory effect to occur within 60 min of exposure (data not shown).

Figure 2. The effect of acetylchoine (ACh) stimulation on neutrophil chemotaxis. Neutrophils were treated with varied concentrations of ACh for 60 min prior to exposure to rhIL-8 (closed diamond) or complement activated serum (open diamond). Neutrophil chemotactic activity (NCA) is on the ordinate and the concentration of ACh is on the abscissa. Values are expressed as means ± SEM. For each experiment a significant dose effect was demonstrated (ANOVA, p < 0.0001; 15 observations per experiment). *p < 0.05, **p < 0.001 means differed as compared with those from non-treated controls.
Tiotropium pretreatment inhibited cholinergic stimulation of neutrophils
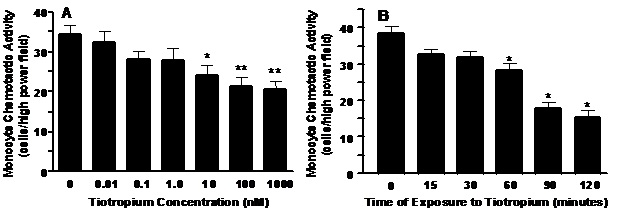
Neutrophils were pretreated for 30 min with varied concentrations of tiotropium ranging between 0.1 to 1000 nM prior to exposure to ACh. Tiotropium pretreatment significantly reversed the stimulatory effect of effect of ACh on NCA at concentrations ranging greater than 1 nM. A dose dependent was observed with the maximum reduction approaching 45% (p<0.001) at levels beyond 10 nM (Figure 3).

Figure 3. The effect of tiotropium on ACh-stimulated neutrophil chemotaxis. Neutrophils were treated with tiotropium at various concentrations (0.1 to 1000 nM) for 30 min prior to treatment to ACh for an additional 60 min and exposure to rhIL-8 as the chemoattractant. Values are expressed as means ± SEM. A treatment effect was demonstrated by one-way ANOVA (p < 0.0001) for three independent experiments. #p < 0.01 means differed compared with non-treated controls; *p < 0.05, **p < 0.001 means differed compared with those from ACh-stimulated neutrophils.
The effect of selective muscarinic receptor antagonists on cholinergic stimulation of neutrophil chemotaxis.
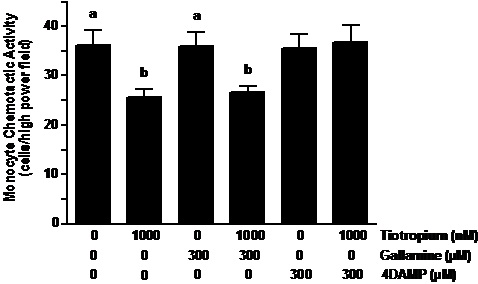
It has been recently demonstrated that neutrophils express muscarinic receptors sub-types 1 through 3 (10,11) and tiotropium can interact amongst these receptors as an antagonist with varying affinities (M3>M1>M2) (1). Thus, we examined the effect of a variety of muscarinic receptor antagonists with specificity to the varied receptors including pirenzepine dihydrochloride (M1), gallamine trithiodide (M2) and 4-DAMP (M3). Neutrophils were pretreated with these muscarinic antagonists at the varied concentrations (0.1 – 1000 nM) for 30 min prior to exposure to 100 µM ACh.
As demonstrated in Figure 4C, 4-DAMP significantly inhibited the increase of NCA that resulted from ACh treatment (32% decrease; p<0.05) although this effect was not as robust as those of tiotropium demonstrated in Figure 3. In contrast to 4-DAMP, gallamine pretreatment did not alter NCA that was stimulated by ACh treatment. Although not significant, an inhibitory trend was observed by pirenzepine pretreatment on cholinergic stimulation of NCA (Figure 4A).

Figure 4. The effect of various muscarinic (M) receptor antagonists on ACh-stimulated neutrophil chemotaxis. Neutrophils were treated with pirenzepine (M1 inhibitor; figure 3A), gallamine (M2 inhibitor; figure 3B) and 4-DAMP (M3 inhibitor; figure 3C) at various concentrations (0.1 – 1000 nM) for 30 min prior to treatment with ACh and exposure to rhIL-8. Values are expressed as means ± SEM. Treatment effects were displayed by ANOVA for pirenzepine (p < 0.03; n = 5), gallamine (p < 0.02; n = 3) and 4-DAMP (p < 0.01; n = 3) experiments. #p < 0.05 means differed as compared with non-treated controls. *p< 0.05 means differed compared with those from ACh-stimulated cells.
Tiotropium bromide effects NCA by altering M3 receptor Gq-protein coupling
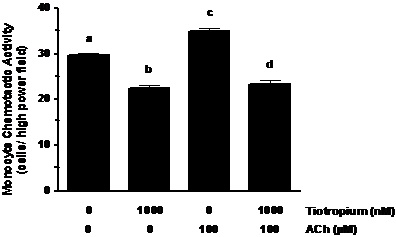
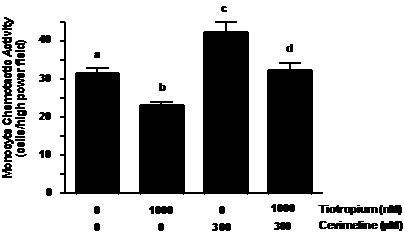
As suggested by results of the muscarinic receptor antagonists above, the M3 receptor seems to play a prominent role in the regulation of cholinergic induction of NCA. To confirm this role, we examined the effect of the specific M3 receptor agonist cevimeline on NCA. Neutrophils were pretreated with tiotropium (30 min) prior to exposure to 300 µM cevimeline for an additional 30 min. As seen in Figure 4A, NCA was promoted by cevimeline treatment when exposed to rhIL-8 (~41% increase as compared to controls; p < 0.001). Similar to the response demonstrated in the ACh series of experiments, tiotropium pre-treatment significantly reversed the stimulatory effect of cevimeline (~40% decrease, p < 0.001) on NCA to a level that was similar to non-treated control neutrophils (Figure 5A).

Figure 5. Tiotropium inhibited the stimulatory effect of cevimeline (M3 receptor agonist) and pasteurella multocida toxin (PMT; Gq signaling stimulator) on NCA. Neutrophils were pre-treated with tiotropium for 30 min before the addition of cevimeline (Figure 4A) or PMT (Figure 4B) for an additional 30 min and exposure to rhIL-8. Values are expressed as means ± SEM. Treatment effects were displayed for both series of experiments (ANOVA; p < 0.0001, n=3). #p < 0.001 means differed as compared with non-treated neutrophils. *p < 0.001 means differed as compared with those from cevimeline- and PMT- stimulated cells.
G-proteins are important regulators in chemokine and complement mediated chemotaxis, and are early-stage regulatory components coupled to muscarinic receptor function (19,24,25). To test whether M3 receptor coupled G-protein pathway is involved in the regulation of cholinergic stimulation of NCA, we treated neutrophils with a potent Gq agonist (Pasteurella multocida toxin; PMT) (26). As demonstrated in Figure 5B, PMT treatment effectively stimulated NCA (~32% increase; p < 0.001) when compared to non-treated controls. In addition, when the neutrophils were pretreated with tiotropium for 30 min prior to PMT stimulation, NCA was markedly reduced by 38% (p < 0.001) as compared to PMT treatment alone (Figure 5B). To further examine the specificity of this event we treated neutrophils with mastoparan, an agonist of the Go proteins coupled to the M2 and M4 receptor function. In contrast to PMT, mastoparan treatment did not influence NCA (data not shown).
Cholinergic activation of ERK-1/2 in neutrophils is inhibited by tiotropium treatment.
It has been previously established that ERK-1/2 protein activation provides a pivotal regulatory role on neutrophil chemotaxis (22,27,28) and it is a downstream signaling pathway that is influenced by G-proteins (29-31). Thus, we examined the effect of ACh activation (100 µM) on ERK-1/2 signaling in neutrophils and began with examining the effect of time of cholinergic exposure (0 to 60 min) on ERK-1/2 protein expression. As seen in Figure 5, ACh treatment activated pERK-1/2 expression but did not alter the level of tERK-1/2 proteins in the cells. The stimulation of pERK-1/2 reached the maximal effect within 15-30 min of exposure to ACh, and began to decrease after 45 min of exposure (Fig 6). Similar reductions on pERK-1/2 expression were demonstrated in experiments where neutrophils were treated with ACh for longer periods (>60 min; data not shown).

Figure 6. The effect of time of exposure of ACh on ERK-1/2 protein activation in neutrophils. Cells were treated with 100 μM ACh for various times from 0 through 45 min of exposure. Total cell proteins were isolated and examined for phosphorylated (p) and total (t) ERK-1/2 expression assessed by Western-blot (Figure 5A). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was assessed as a loading control. The corresponding mean ratio of pERK-1/2: tERK-1/2 resulting from densitometric scans is demonstrated in figure 6B.
A further series of experiments (n=4) were conducted to examine the effect of tiotropium on the inhibition of cholinergic stimulation of ERK-1/2. Neutrophils were pretreated with 100 nM tiotropium for 30 min prior to exposure to 100 µM ACh for 15 min. As seen in Figure 7, ACh treatment increased the activation of ERK proteins (pERK/tERK ratio = 0.85 for ACh vs. 0.52 in non-treated control cells; p < 0.01) and tiotropium pretreatment markedly reversed this effect and where expression was reduced to control cell levels (Figure 7).

Figure 7. The effect of tiotropium on ACh stimulated ERK-1/2 protein activation in neutrophils. Cells were treated with 100 nM of tiotropium prior to expose to 100 μM of ACh for 15 min. Total cell proteins were isolated and pERK-1/2, tERK-1/2 and GAPDH expression was determined by Western-blot. A representative experiment is shown in Figure 6A and ERK-1/2 activation expressed as pERK1/2: tERK-1/2 is demonstrated in Figure 6B. Values are expressed as means ± SEM. A treatment effect was demonstrated by ANOVA (p < 0.005; n=4 experiments). #p < 0.01 means differ as compared with non-treated neutrophils; * p < 0.01 means differ as compared to those from ACh stimulated cells.
Cholinergic stimulation of NCA is reduced by an inhibitor of ERK-1/2 phosphorylation
Based of the aforementioned results on pERK-1/2 expression activation of ERK-1/2 by phosphorylation may govern NCA. Neutrophils were pretreated with U0126 (a specific MEK-1/2 kinase inhibitor) for 30 min prior to exposure to ACh (100µM) for an additional 60 min. As seen in Figure 8, U0126 pretreatment strongly inhibited (p < 0.01) the increase of NCA by ACh treatment to levels similar to non-treated control cells.

Figure 8. Neutrophil chemotactic activity that is stimulated by ACh is inhibited by an antagonist of ERK-1/2 phosphorylation. Cells were pretreated with a specific inhibitor of MEK-1 and -2 (U0126; 10 µM) for 30 min prior to the addition of 100 µM ACh for an additional 30 min before assessing NCA as described above. Values are expressed as means ± SEM. A treatment effect was demonstrated by ANOVA (p < 0.001) resulting from three independent experiments. #p < 0.001 means differed as compared with non-treated neutrophils. *p< 0.01 means differed compared with those from ACh stimulated cells.
Discussion
Previous clinical results have suggested that tiotropium inhalation provides beneficial clinical outcomes in COPD that may result from modulating mechanisms beyond bronchodilatation (1,10). An intriguing suggestion has been that anticholinergic therapy may act as an anti-inflammatory. The mechanism(s) by which this action occurs has not been fully elucidated; however, recent in vitro studies have suggested that tiotropium may indirectly influence neutrophil chemotaxis by inhibiting the release of chemotactic factors by resident lung cells that would subsequently promote NCA (3,4). In a model using U937 macrophages, we previously reported that NCA was decreased from supernatants that were obtained from LPS-challenged cells treated with tiotropium and that this result did not occur from a reduction in corresponding chemotactic factor expression measured in the supernatants (17). Specifically, we found that heightened levels of IL-8 did not correlate (r = 0.38; p > 0.13) with the reduction in NCA upon tiotropium treatment (0.1 to 1000 nM). Similar effects were also shown regarding LTB4 analyses (17). Based on these results, we formulated the hypothesis that tiotropium contained in the supernatants may actually interact with the neutrophils and influence their activity directly.
Current concepts suggest that an influx of neutrophils is important in the pathogenesis of COPD (18). These neutrophils release proteases and toxic oxygen radicals that contribute to the inflammation seen in COPD. It is this inflammation that results in the emphysema and airway remodeling that causes the structural changes in COPD that lead to the clinical symptoms of breathlessness and/or cough. Previous studies in animal models of COPD have shown that tiotropium is anti-inflammatory (5,32). More recent studies in humans suggest that tiotropium reduces neutrophil chemotaxis (33). Migration of neutrophils from COPD patients are also decreased by tiotropium similarly to the normal human neutrophils used in this study (34). The present studies are consistent with these results and support an anti-inflammatory role for tiotropium in COPD.
It has not been established to date that cholinergic stimulation may directly affect NCA in vitro. In the present study, we report that exogenous ACh pretreatment enhanced NCA when the cells were exposed to differing chemotactic agents. In addition, we found that tiotropium treatment prior to ACh exposure very effectively reduced stimulated NCA. The bioactive concentrations of tiotropium that were used in this study initially ranged from 10 -1000 nM and the lower bioactive responsive doses were similar to those previously reported to affect human lung fibroblast proliferation (35), fibroblast differentiation (16), and the release of chemotactic factors from epithelial cells, fibroblasts and alveolar macrophages in vitro (3,4). In order to elicit a robust effect on NCA, we opted to use a dose of 100 nM of tiotropium throughout the study. At this level, tiotropium was non-toxic and remained below the estimated maximum concentration of ~2000 nM to be present in the lung epithelial lining fluid after inhalation of the drug (36).
There is increasing evidence that signaling from extraneuronally produced ACh may play an important role in regulation of lung inflammation (1,9). ACh may enhance proinflammatory cell chemotaxis indirectly by stimulating resident lung cells to release chemotactic factors (3,4,14,15). Recently, choline acetyltranferase (ChAT) has been localized in human blood and skin derived neutrophils; however, to date there have been no studies establishing ChAT expression in pulmonary neutrophils (10). However, a recent report by Neumann et al. (37) demonstrated that mononuclear cells (T cells and monocytes) expressed ~0.36 pmol ACh/106 cells, whereas granulocytes (containing predominantly neutrophils) expressed considerably less concentration of ACh although their synthetic capacity was greater than CD3+ cells. Thus, it remains to be established whether pulmonary neutrophils may produce Ach, especially under conditions of inflammation. It also remains to be established whether neutrophils produce sufficient ACh to regulate a cholinergic response in an autocrine manner.
It has also been reported that neutrophils express muscarinic receptors (9.10,13,38). Interestingly, the expression of muscarinic receptors is modulated in neutrophils in COPD, particularly the M3 receptors which are more highly expressed under this condition (13). In this study we demonstrated that neutrophils may react to exogenous cholinergic stimulation thus suggesting that paracrine cholinergic stimulation may be a viable mechanism of control of neutrophil activity associated with inflammation.
In an early attempt to characterize the muscarinic receptor(s) involved in cholinergic regulation of NCA we used a panel of selective antagonists and tested their reactivity against ACh stimulation. To accomplish this objective we pretreated neutrophils with pirenzepine, gallamine and 4-DAMP prior to cholinergic treatment. Our results demonstrated that only the inhibitor 4-DAMP significantly reversed the effect of ACh on NCA. These results further confirm that anti-inflammatory control may entail the antagonism of the muscarinic type-3 receptor. This is comparable to our previous studies that have demonstrated that ACh may promote chemotactic factor release from resident lung cells by influencing M3 receptors (4,14,15).
We further treated neutrophils with cevimeline, a M3 receptor agonist (39), and found that this compound markedly increased NCA. When neutrophils were treated with PMT, a Gq agonist (27,40), it stimulated their activity and to a level comparable to those of cevimeline. Moreover, tiotropium pretreatment dramatically inhibited PMT stimulated NCA. Taken together, these results suggest that tiotropium may interact with the M3 receptor and possibly modulate early Gq mediated signaling cues affecting NCA by cholinergic treatment.
The M3 receptors have the capacity to activate multiple signaling pathways in various cell types. For example, it has been established that the M3 receptor and Gq protein pathway is involved in airway smooth muscle contraction and may function by regulating PLC, inositol 1,4,5-triphosphate (IP3) and intracellular Ca2+ signaling (41). In addition, it has been shown that Gq-deficient neutrophils possess deficient calcium signaling and defective chemotactic responsiveness (42). Furthermore it has been reported that ERK activation is associated with Gq-protein stimulation (29,30) and ERK signaling is an important integral regulator of NCA (22,27,28). In this study, we find that ACh treatment enhanced neutrophil ERK-1/2 protein phosphorylation but not total ERK1/2 expression. In addition, the pretreatment of the cells with tiotropium reversed this activity. Similarly, Profita et al. (4) demonstrated that ACh mediated release of IL-8 in human bronchial epithelial cells may be regulated in part by an ERK-dependent mechanism.
In summary, these data support the role of cholinergic stimulation on NCA an important inflammatory process contributing to pulmonary disease. This study also demonstrated an alternative anti-inflammatory role of tiotropium on directly reducing chemotactic activity by inhibiting, in part, Gq protein and ERK activation in neutrophils. Furthermore, this action was independent of type or concentration of chemotactic factor. This present study may provide some insight into the recently reported discordance between significant reductions in total exacerbation compared with no reduction in proinflammatory marker (including IL-8) concentration in sputa from COPD patients treated with tiotropium (43). The inhibition of neutrophil migration is one effect which may contribute to the anti-inflammatory effects of anticholinergics and may explain, at least in part, the reduction in exacerbations of COPD seen with tiotropium.
Acknowledgements
This study was funded by Boehringer Ingelheim and the Phoenix Pulmonary and Critical Care Research and Education Foundation and the Department of Veterans Affairs. The contents do not represent the views of the Department of Veterans Affairs or the United States Government..
References
- Restrepo RD. Use of inhaled anticholinergic agents in obstructive airway disease. Respir Care 2007;52:833-851.
- Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to emphysema. New Engl J Med 1984;311:421-425.
- Bühling F, Lieder N, Kühlmann UC, Waldburg N, Welte T. Tiotropium suppresses acetylcholine-induced release of chemotactic mediators in vitro. Respir Med 2007;101:2386-94.
- Profita M, Bonanno A, Siena L, Ferraro M, Montalbano AM, Pompeo F, Riccobono L, Pieper MP, Gjomarkaj M. Acetylcholine mediates the release of IL-8 in human bronchial epithelial cells by a NFkB/ERK-dependent mechanism. Eur J Pharmacol 2008;582:145-53.
- Wollin L, Pieper MP. Tiotropium bromide exerts anti-inflammatory activity in a cigarette smoke mouse model of COPD. Pulm Pharmacol Ther 2010;23:345-54.
- Profita M, Bonanno A, Montalbano AM, Ferraro M, Siena L, Bruno A, Girbino S, Albano GD, Casarosa P, Pieper MP, Gjomarkaj M. Cigarette smoke extract activates human bronchial epithelial cells affecting non-neuronal cholinergic system signaling in vitro. Life Sci 2011;89:36-43.
- Profita M, Riccobono L, Montalbano AM, Bonanno A, Ferraro M, Albano GD, Gerbino S, Casarosa P, Pieper MP, Gjomarkaj M. In vitro anticholinergic drugs affect CD8+ peripheral blood T-cells apoptosis in COPD. Immunobiology 2012;217:345-53.
- Profita M, Bonanno A, Montalbano AM, Albano GD, Riccobono L, Siena L, Ferraro M, Casarosa P, Pieper MP, Gjomarkaj M. β(2) long-acting and anticholinergic drugs control TGF-β1-mediated neutrophilic inflammation in COPD. Biochim Biophys Acta 2012;1822:1079-89.
- Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respir Res 2006;7:73.
- Gwilt CR, Donnelly LE, Rogers DF. The non-neuronal cholinergic system in the airways: an unappreciated regulatory role in pulmonary inflammation? Pharmacol Ther 2007;115:208-222.
- Racke K, Juergens UR, Matthiesen S. Control by cholinergic mechanisms. Eur J Pharmacol 2006;533:57-68.
- Wessler IK, Kirkpatrick CJ. The non-neuronal cholinergic system: an emerging drug target in the airways. Pulm Pharmacol Therap 2001;14:423-434.
- Profita M, Giorgi RD, Sala A, Bonanno A, Riccobono L, Mirabella F, Gjomarkaj M, Bonsignore G, Bousquet J, Vignola AM. Muscarinic receptors, leukotriene B4 production and neutrophilic inflammation in COPD patients. Allergy 2005;60:1361-1369.
- Koyama S, Rennard SI, Robbins RA. Acetylcholine stimulates bronchial epithelial cells to release neutrophil and monocyte chemotactic activity. Am J Physiol 1992;262:L466-471.
- Sato E, Koyama S, Okubo Y, Kubo K, Sekiguchi M. Acetylcholine stimulates alveolar macrophages to release inflammatory cell chemotactic activity. Am J Physiol 1998;274:L970-979.
- Pieper MP, Chaudhary NI, Park JE. Acetylcholine-induced proliferation of fibroblasts and myofibroblasts in vitro is inhibited by tiotropium bromide. Life Sci 2007;80:2270-2273.
- Rempe S, Robbins RA, Hoyt JC, Kurai M, Koyama S, Hayden JM. Tiotropium inhibits neutrophil chemotaxis [abstract]. Am J Resp Crit Care Med 2007;175:A493.
- Niggli V. Signaling to migration in neutrophils: importance of localized pathways. Int J Biochem Cell Biol 2003;35:1619-1638.
- Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 2008;8:183-92.
- Ottonello L, Montecucco F, Bertolotto M, Arduino N, Mancini M, Corcione A, Pistoia V, Dallegri F. CCL3 (MIP-1alpha) induces in vitro migration of GM-CSF-primed human neutrophils via CCR5-dependent activation of ERK 1/2. Cell Signal 2005;17:355-63.
- Fuhler GM, Knol GJ, Drayer AL, Vellenga E. Impaired interleukin-8- and GROalpha-induced phosphorylation of extracellular signal-regulated kinase result in decreased migration of neutrophils from patients with myelodysplasia. J Leukoc Biol 2005;77:257-66.
- Hii CS, Anson DS, Costabile M, Mukaro V, Dunning K, Ferrante A.Characterization of the MEK5-ERK5 module in human neutrophils and its relationship to ERK1/ERK2 in the chemotactic response. J Biol Chem 2004;279:49825-34.
- Böyum A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest 1968;97:77-89.
- Honda Z, Takano T, Hirose N, Suzuki T, Muto A, Kume S, Mikoshiba K, Itoh K, Shimizu T. Gq pathway desensitizes chemotactic receptor-induced calcium signaling via inositol trisphosphate receptor down-regulation. J Biol Chem 1995;270:4840-4844.
- Wess J. Molecular basis of receptor/G-protein-coupling selectivity. Pharmacol Therap 1998;80:231-64.
- Wilson BA, Ho M. Pasteurella multocida toxin as a tool for studying Gq signal transduction. Rev Physiol Biochem Pharmacol 2004;152:93-109.
- Van Lint J, Van Damme J, Billiau A, Merlevede W, Vandenheede JR. Interleukin-8 activates microtubule-associated protein 2 kinase (ERK1) in human neutrophils. Mol Cell Biochem 1993;127-128:171-7.
- Coxon PY, Rane MJ, Uriarte S, Powell DW, Singh S, Butt W, Chen Q, McLeish KR. MAPK-activated protein kinase-2 participates in p38 MAPK-dependent and ERK-dependent functions in human neutrophils. Cell Signal 2003;15:993-1001.
- Budd DC, Rae A, Tobin AB. Activation of the mitogen-activated protein kinase pathway by a Gq/11-coupled muscarinic receptor is independent of receptor internalization.J Biol Chem 1999;274:12355-60.
- Budd DC, Willars GB, McDonald JE, Tobin AB. Phosphorylation of the Gq/11-coupled m3-muscarinic receptor is involved in receptor activation of the ERK-1/2 mitogen-activated protein kinase pathway. J Biol Chem 2001;276:4581-7.
- Osmond RI, Sheehan A, Borowicz R, Barnett E, Harvey G, Turner C, Brown A, Crouch MF, Dyer AR.GPCR screening via ERK 1/2: a novel platform for screening G protein-coupled receptors. J Biomol Screen 2005;10:730-7.
- Pera T, Zuidhof A, Valadas J, Smit M, Schoemaker RG, Gosens R, Maarsingh H, Zaagsma J, Meurs H. Tiotropium inhibits pulmonary inflammation and remodelling in a guinea pig model of COPD. Eur Respir J 2011;38:789-96.
- Vacca G, Randerath WJ, Gillissen A. Inhibition of granulocyte migration by tiotropium bromide. Respir Res 2011;12:24.
- Santus P, Buccellati C, Centanni S, Fumagalli F, Busatto P, Blasi F, Sala A. Bronchodilators modulate inflammation in chronic obstructive pulmonary disease subjects. Pharmacol Res. 2012;66:343-8.
- Matthiesen S, Bahulayan A, Kempkens S, Haag S, Fuhrmann M, Stichnote C, Juergens UR, Racke K. Muscarinic receptors mediate stimulation of human lung fibroblast proliferation. Am J Respir Cell Mol Biol 2006;35:621-7.
- Disse B, Speck GA, Rominger KL, Witek TJ, Jr., Hammer R. Tiotropium (Spiriva): mechanistical considerations and clinical profile in obstructive lung disease. Life Sci 1999;64:457-64.
- Neumann S, Razen M, Habermehl P, Meyer CU, Zepp F, Kirkpatrick CJ, Wessler I. The non-neuronal cholinergic system in peripheral blood cells: effects of nicotinic and muscarinic receptor antagonists on phagocytosis, respiratory burst and migration. Life Sci 2007;80:2361-4.
- Bany U, Gajewski M, Ksiezopolska-Pietrzak K, Jozwicka M, Klimczak E, Ryzewski J, Chwalinska-Sadowska H, Maslinski W. Expression of mRNA encoding muscarinic receptor subtypes in neutrophils of patients with rheumatoid arthritis. Ann NY Acad Sci 1999;876:301-304.
- Weber J, Keating GM. Cevimeline. Drugs 2008;68:1691-8.
- Orth JH, Lang S, Taniguchi M, Aktories K. Pasteurella multocida toxin-induced activation of RhoA is mediated via two families of G{alpha} proteins, G{alpha}q and G{alpha}12/13. J Biol Chem 2005;280:36701-7.
- McGraw DW, Elwing JM, Fogel KM, Wang WC, Glinka CB, Mihlbachler KA, Rothenberg ME, Liggett SB. Crosstalk between Gi and Gq/Gs pathways in airway smooth muscle regulates bronchial contractility and relaxation. J Clin Invest 2007;117:1391-8.
- Shi G, Partida-Sánchez S, Misra RS, Tighe M, Borchers MT, Lee JJ, Simon MI, Lund FE. Identification of an alternative G{alpha}q-dependent chemokine receptor signal transduction pathway in dendritic cells and granulocytes. J Exp Med 2007;204:2705-18.
- Powrie DJ, Wilkinson TM, Donaldson GC, Jones P, Scrine K, Viel K, Kesten S, Wedzicha JA. Effect of tiotropium on sputum and serum inflammatory markers and exacerbations in COPD. Eur Respir J 2007;30:472-8.
Reference as: Kurai M, Robbins RA, Koyama S, Amano J, Hayden JM. Acetylcholine stimulation of human neutrophil chemotactic activity is directly inhibited by tiotropium involving Gq and ERK-1/2 regulation. Southwest J Pulm Crit Care 2012:5:152-68. (Click here for a PDF version)


 Post a Comment
Post a Comment